We have released a new website. Please give it a try and let us know your thoughts.
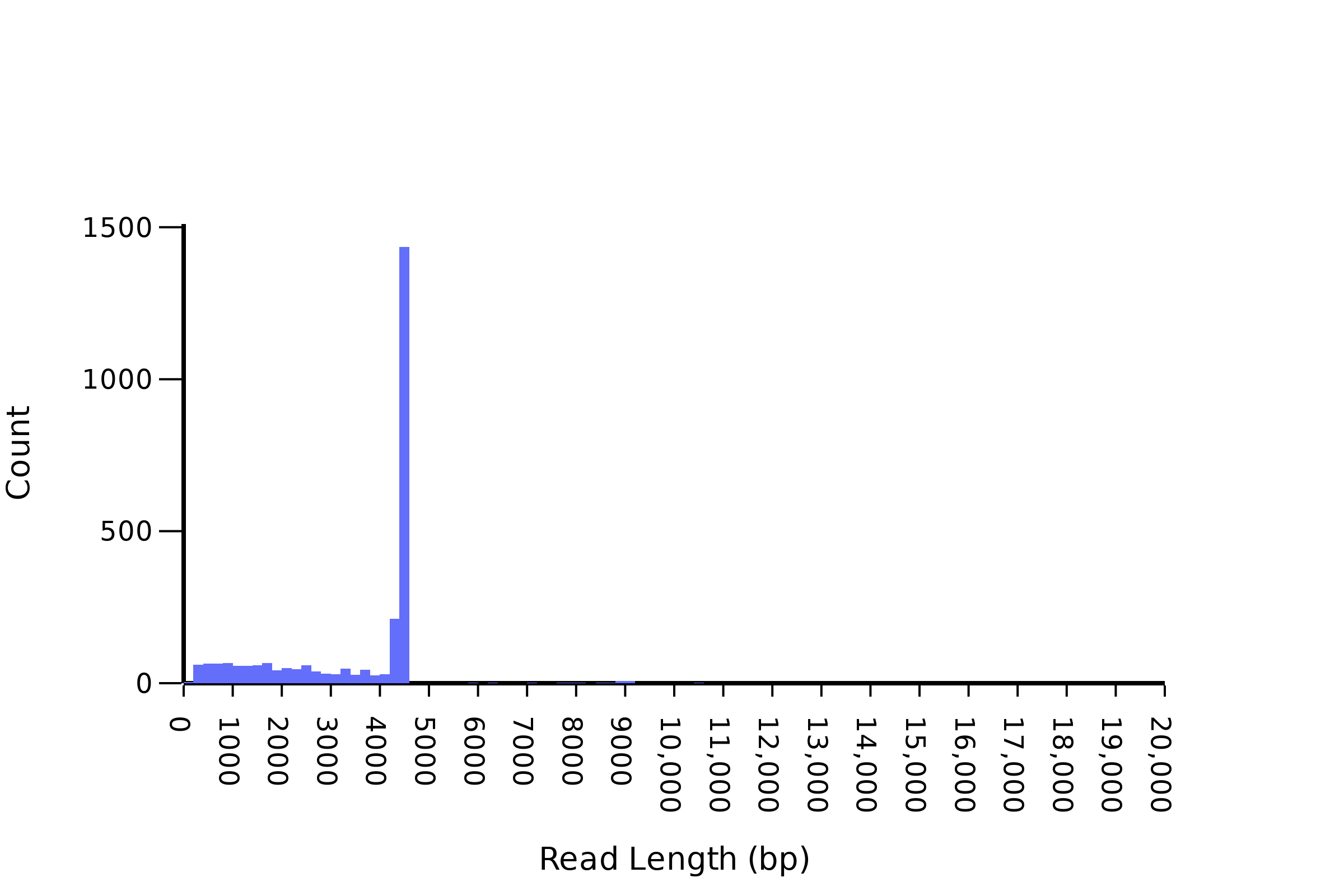
Sometimes we find that there are no plasmid reads that correspond with the size that you expect. This can be because your cloning has not worked and you have an empty backbone, however, we also commonly see plasmid concatamers and mixed samples. The best way to troubleshoot this is to look at the _length.png file that we provide. If you have a single plasmid you expect to see the below graph with a single peak and minimal genomic DNA contamination.
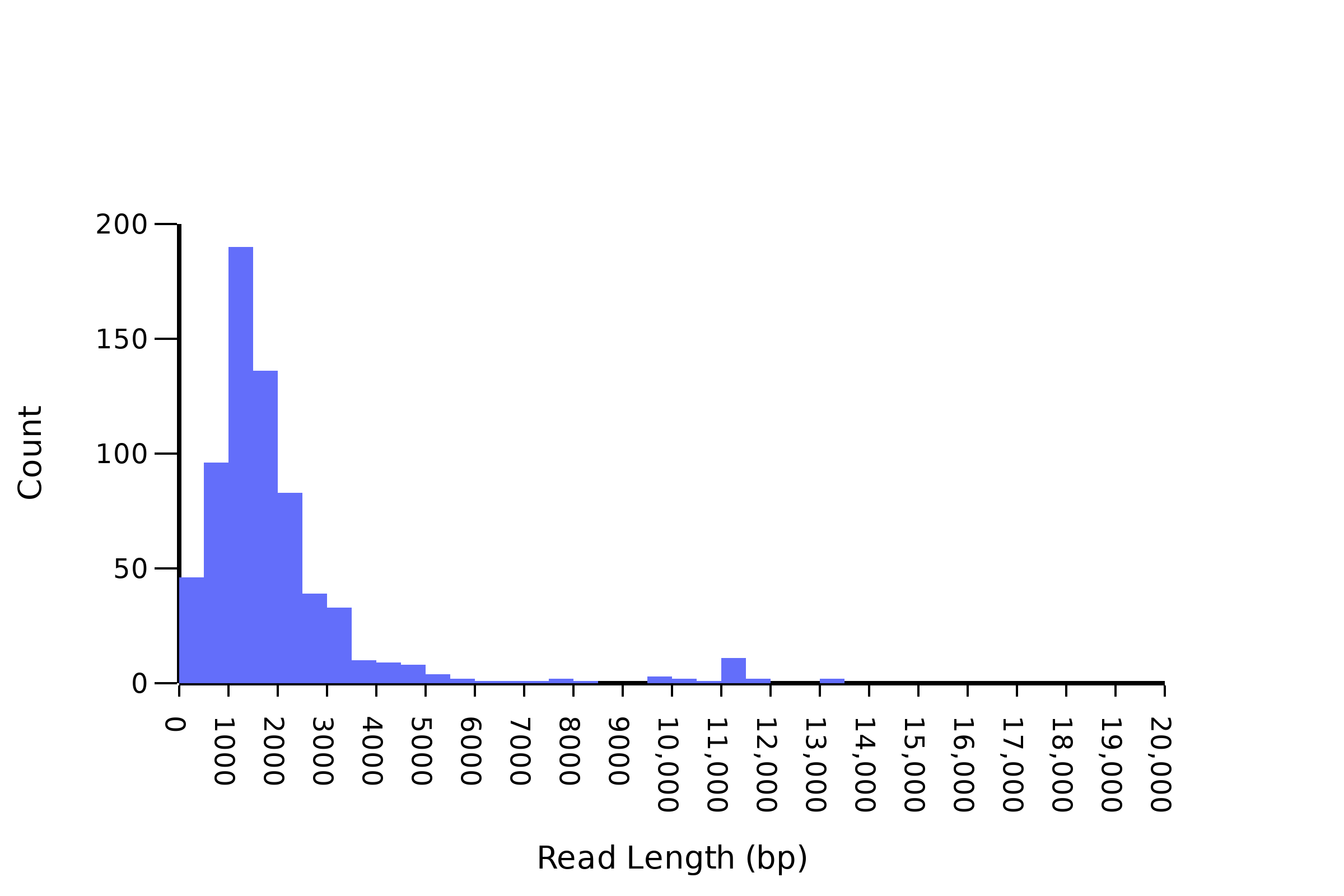
The most common failure modes for nanopore whole plasmid sequencing are (1) genomic DNA contamination and (2) too low sample concentration. We often find that there is substantial genomic DNA contamination and this essentially leads to low effective plasmid concentrations. Although other providers recommend sending Qubit quantified DNA we find that this can be quite the ask for most groups. We ask that you run an electrophoresis gel when you first send us samples to check the quality of your plasmid preparations and ensure the spectrophotometrically measured DNA concentrations make sense given what you see on your gel. Genomic DNA contamination will look like the graph below in the _length.png plot that we send you.
Nanopore has two "failure modes" when it comes to basecalling. The first is homopolymers regions (polyA, G, T and C) where it often is not able to tell how long the stretch is as the current doesn't change while the strand moves through the pore. The second is methylated regions where although there are basecalling models to call base modifications
